


 >
新闻
>
新闻
点击量:221 时间:2021-07-19
近年来,靶向蛋白降解嵌合体 (PROTAC)作为新颖的诱导蛋白降解方式已成为一种全新的药物发现策略。来自英国人工智能公司BenevolentAI的Ian Churcher从药物发现的角度对PROTAC历史、技术的独特优势和面临的挑战进行了综述。

传统药物发现策略的局限性
传统药物设计常常专注于优化药物结合亲和力,但往往限制了更多高效药物的发现,因为识别一种高效能和高选择性的药物来调节一个生物靶点并不总是那么简单,面对越来越多缺乏高亲和力配体结合位点的药物干预靶点的发现,传统的小分子成药技术显得无计可施。
越来越多的方法被用来规避这些限制,单克隆抗体和其他蛋白制剂虽然具有高亲和力和选择性的优势,但这些制剂目前只能应用于细胞外或细胞表面靶点,而大多数基因组蛋白质和当代药物靶点是在细胞内发挥作用。通过基因沉默或RNA抑制剂作用于靶点,虽然在临床前是非常有效的,但必需的寡核苷酸制剂本身也存在着多项挑战(细胞传递、稳定性、生物分布、选择性等),必须克服这些挑战才能开发出成功的药物。
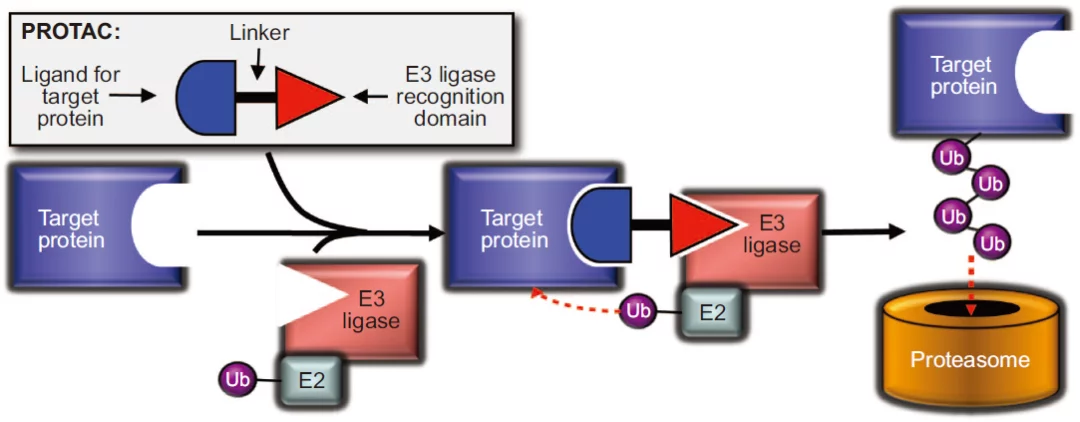
图1. PROTAC诱导蛋白降解的作用机制
图片来源:ACS Chemical Biology
靶向蛋白降解嵌合体 (PROTAC)技术简介
靶向蛋白降解嵌合体 (PROTAC)不同于传统的小分子抑制剂,PROTAC采用的是事件驱动(event-driven)的药理学作用模式,而非传统的小分子占据驱动模式(occupancy-driven)。PROTAC诱导蛋白降解的作用机制见图1,PROTAC分子包括三部分,一头是靶向目的蛋白结构,另一头招募泛素E3连接酶,中间通过合适的连接体(linker)连接,通过连接体将靶蛋白配体与泛素E3连接酶配体连接一起,在体内分别识别靶蛋白和泛素E3连接酶,通过诱导招募泛素E3连接酶至靶蛋白表面,引发多聚泛素化过程诱导靶蛋白降解,从而达到疾病治疗的效果。这种全新的小分子诱导药理学的途径对药物发现过程产生了深远影响。
借用自然:PROTAC技术的起源
自然界已经发展出一系列的生物途径,通过精准的信号增强和衰减以及其他过程来维持生物内环境的稳定和细胞对刺激的反应。这些途径只有一小部分步骤使用直接的小分子调节器(如激素和其他GPCR配体、突触信号、代谢物传感和调节),大多数调节是通过复杂的途径进行的,最终导致蛋白质合成或转换。随着我们对细胞内源性蛋白质降解机制的理解越来越清楚,潜在的治疗策略也开始出现。细胞内蛋白质的降解主要有两种途径,即泛素-蛋白酶体系统(UPS)和自噬/溶酶体途径。其中,泛素-蛋白酶体系统是细胞内蛋白质降解的主要途径,参与细胞内80%以上。以色列科学家Aaron Ciechanover、Avram Hershko和美国科学家Irwin Rose因共同发现了泛素调节的蛋白降解过程,获得了2004年的诺贝尔化学奖。PROTAC技术便是来源于这项诺贝尔奖,其在一定程度上利用细胞内的泛素-蛋白酶体系统来降解掉目的蛋白,PROTAC将靶点蛋白连接到泛素E3连接酶,招募E2连接酶给目标蛋白加上泛素标签,多聚泛素化之后的蛋白质被蛋白酶体识别并被蛋白质水解为氨基酸,PROTAC分子释放出来,继续进行重复的过程(图2)。
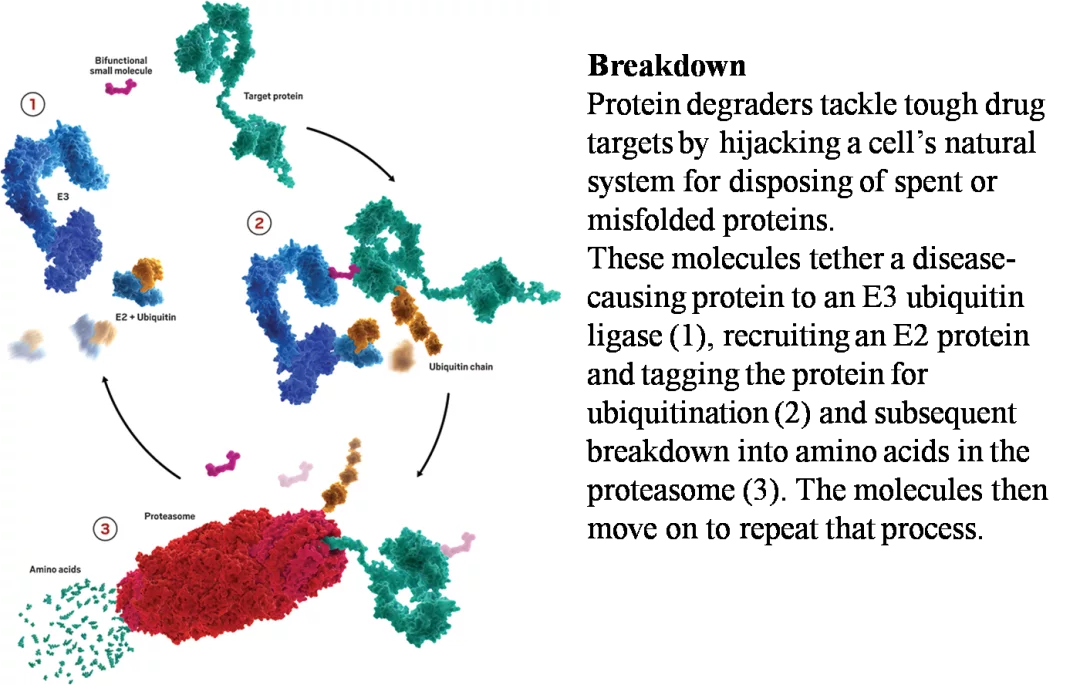
图2. 靶点蛋白水解过程-图片来源:Kymera Therapeutics
在设计PROTAC分子时,需要特别关注以下几点,PROTAC分子必须与靶点蛋白和泛素化机制结合,形成有效的三元复合物,才能实现靶蛋白降解。因此,首先要保障PROTAC能透过细胞膜成功的进入细胞;一旦进入细胞内,蛋白质降解分子必须同时与目标蛋白和泛素化机制结合,形成三元复合物,避免形成二元复合物“hook effect”;三元复合物一旦形成,泛素必须以足够的速率(快于三元复合物的本征寿命)转移到靶蛋白受体位点上(通常是表面赖氨酸),所需底物的诱导泛素化也应以这样的速率进行,以克服由二氢奎素酶竞争性去除泛素的情况;最后,泛素转移到底物蛋白上的模式应允许蛋白酶体容易识别,从而开始实际降解。
蛋白质可与多种泛素形成不同长度的链和不同赖氨酸残基(例如K6、K11、K27、K29、K33、K48、K63和M1)连接的拓扑结构。在生理学上,不同的多泛素连接和链长/拓扑结构可以导致在蛋白质功能精细调节的复杂系统中的一系列细胞反应。其中,K48连接泛素和其他的线性链被认为有利于蛋白酶体识别。另外,蛋白酶体起始区域部分无序loop或domain的存在也很重要,在某些情况下,如果该区域不存在,蛋白质虽然可以广泛多聚化,但不被蛋白酶体识别。
从概念到实践:PROTAC技术的发展历程
回顾PROTAC技术20年的发展历史,有很多学者做出了重要贡献,一些里程碑事件助力了PROTAC技术的发展,接下来将简述下PROTAC技术的发展历程(图3)。人工诱导蛋白质降解的最早报道之一是在1995年设计了一系列与Ig结合基序融合的修饰E2结合酶(如TaUBC4),首次表明人工诱导的泛素化特征可被诱导降解的蛋白酶体识别。
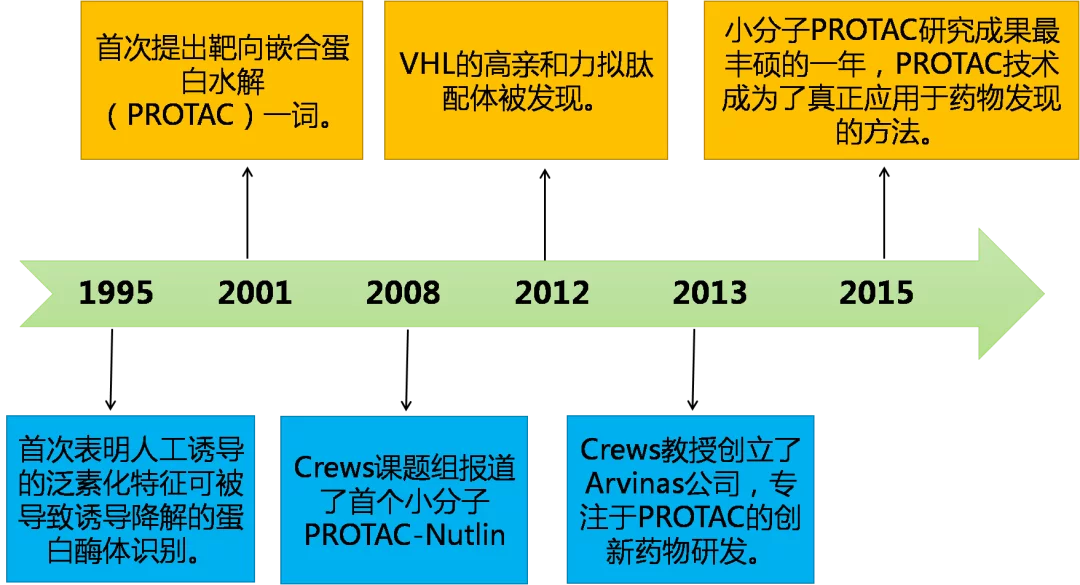
图3. PROTAC领域发展史
2001年, Deshaies和Crews实验室利用了含SCFβ-TRCP的E3诱导了MetAp-2的降解,首次引入了靶向嵌合蛋白水解(PROTAC)一词。这篇早期开创性论文选择了一种卵白蛋白衍生物,它通过共价相互作用与Met-AP2结合。这种对共价键的依赖阻止了PROTAC的独特优点之一,即其催化作用(见下文),潜在地限制了所观察到的降解效率。在接下来的几年里, PROTAC发展到能够降解诸如雌激素受体(ER)和雄激素受体(AR)等靶点,由于早期的PROTAC分子多为小肽,细胞渗透性比较差,细胞活性也较低,无法应用到药物的开发中。
直到2008年,Crews课题组首次报道了小分子PROTAC-nutlin,典型的小分子PROTAC结构如图4所示,作用机制如图5所示。Nutlin通过与E3连接酶Mdm2结合,诱导雄激素受体(AR)的降解,但其细胞活性仍不理想,在10μM浓度下才诱导AR降解,与此同时,尽管从所报道的有数据来看,降解是蛋白酶体依赖性的,但考虑到许多AR配体可以自发地破坏其同源受体的稳定性,引起自身泛素化和降解。因此,其在药物发现的应用仍然有限。
图4. 小分子PROTAC的三个基本单元,图片来源:JMC

图5. PROTAC分子的作用机制,图片来源:JMC
2010-2012年,随着Craig Crews团队(耶鲁大学的该领域的先驱)和Alessio Ciulli(剑桥大学,E3连接酶结构生物学专家)及其他合作者发现了更多泛素E3连接酶的结合体,PROTAC取得了重大的进展。2012年,VHL的高亲和力拟肽配体被发现。随后,Alessio Ciulli课题组报道了VHL拟肽配体的构效关系研究。
Craig Crews教授在2013年创立了Arvinas公司,公司专注于PROTAC技术,开发创新药物,使得PROTAC技术得以迅速地发展。基于VHL的配体设计的PROTAC化合物具有更多类药特性,可以大幅度提高细胞的穿透性,细胞活性可以达到纳摩尔级别。另外,通过与VHL非结合性对映体设置对照试验证明,PROTAC是通过利用含有VHL的E3泛素连接酶来降解蛋白激酶2(RIPK2)。继前面的研究,Craig Crews团队利用E3泛素连接酶cereblon设计了作用BET家族蛋白的小分子。
2015年是小分子PROTAC研究成果最丰硕的一年,宣布了PROTAC技术成为真正应用于药物发现的方法,吸引了学术界和工业界,以及投资机构的关注,为新药研发打开了新篇章。
PROTAC技术的优势
与传统的小分子拮抗剂相比,PROTAC具有一系列独特的优势:
催化降解功能。传统的小分子采用的药理学作用模式为占据驱动模式,为了提高靶点占有率,往往需要高剂量的药物,反而会带来很大的毒副作用。PROTAC采用的是事件驱动(event-driven)的药理学作用模式,其对目的蛋白的降解过程是一种催化作用,因此只需较低的化合物浓度便可以达到很好的降解效率。
选择性。现代药物开发的一个关键目标是设计高选择性分子,降低药物毒性和副作用。PROTAC分子发挥催化作用时,可以选择性的作用于特定的靶蛋白。最近的多项研究表明, PROTAC对同源靶蛋白的选择性高于传统抑制剂。
作用靶点广。泛素-蛋白酶体系统是细胞内蛋白质降解的主要途径,参与细胞内80%以上。E3泛素连接酶在多种细胞中广泛表达,PROTAC分子只需将靶蛋白与E3泛素连接酶拉近,再通过蛋白溶酶体降解靶蛋白。因此PROTAC技术可以广泛应用不同的靶点。
延长作用时间。靶蛋白的降解是时间依赖性的,PROTAC可以在几分钟内将细胞内靶蛋白消耗到接近基础水平。一旦先前存在的蛋白耗尽,PROTAC只需要降解重新合成的靶蛋白,大多数蛋白质的再合成速度很慢,即使在PROTAC完全清除后,细胞可能仍需要一段相当长的时间,才能将蛋白质库恢复生理信号的水平,从而大大延长作用时间。虽然口服给药通常被认为是小分子药物的首选给药途径,但患者的依从性,特别是对于更慢性的疾病,可能非常差,严重限制了治疗效果。PROTAC可以通过多种途径提供,提供更多有吸引力的临床给药方式。
新的药理学作用模式。许多蛋白靶点对药物研发提出了挑战,它们可能没有一个特定的催化活性位点,需要依赖于更大的蛋白质界面来介导信号,或者蛋白质可能具有多个功能和催化结构域。识别高亲和力抑制剂对于这些靶点来说几乎是一个不可能的事件,但是PROTAC技术可以提供一种解决方案,因为只需要粘合剂(与传统抑制剂相反)来促进E3连接酶的募集和降解级联的启动。PROTAC的另一个优势是其表型基因敲除的能力,利用PROTAC可以作为研究靶基因功能序列丢失的有力补充手段。
展望PROTAC技术的未来
PROTAC技术为新药研发打开了新篇章,也给学术界和工业界带了前所未有的机遇。当然,PROTAC作为一种新兴的技术,必然也会面临很多问题和挑战,只有克服了这些困难,PROTAC技术才能更好的发展。
迄今为止, PROTAC技术已被应用于不同的靶点,从蛋白酶到核激素受体、表观遗传因子和激酶等。目前80%的蛋白缺少现有药物的调控,主要是因为缺少可成药的作用靶点,PROTAC技术应用于不可成药靶点,是未来可以进一步研究和探索的内容。根据人类基因组,科学家预测有超过600个E3泛素连接酶。但迄今为止,只有一小部分被证明适合于PROTAC的开发,为了进一步发展PROTAC技术,未来需要努力确定其他可利用的E3泛素连接酶。设计更有效的PROTAC的另一个关键方面在于连接体Linker,连接体的长度和连接点都可能会影响选择性和降解效率。同时为了简化并加速PROTAC分子的发现,需要建立可靠的PROTAC技术平台。随着PROTAC分子进入临床试验研究,PROTAC技术未来的发展空间是毋容置疑的。
参考文献
Churcher I . Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones?[J]. Journal of Medicinal Chemistry, 2018, 61(2):444-452.
原创 叨叨一下 唯信计算


